La Enfermedad de Von Hippel-Lindau (E VHL) es síndrome hereditario de cáncer familiar que predispone a una variedad de tumores malignos y benignos, como hemangioblastomas localizados en retina, cerebelo, y médula espinal, carcinoma renal de células claras, feocromocitomas o paraganglioma, tumores pancreáticos, tumores del saco endolinfático, cistoadenomas benignos del epidídimo en varones, y tumores del ligamento ancho en mujeres.
¿Cuál es la causa de la VHL?
Toda la información genética está contenida en el núcleo de cada una de nuestras células, en unas estructuras alargadas que son los cromosomas. Tenemos 46 cromosomas, que realmente son dos juegos de 23 cromosomas heredados de cada uno de nuestros padres. A su vez, cuando tenemos hijos, les pasamos a ellos, en cada uno de los espermatozoides u óvulos que producimos.
Cada cromosoma es como una madeja de lana en la que el hilo de la madeja es la cadena de ADN que contiene la información genética. Esta información, almacenada en el código genético, consiste en instrucciones para la producción de proteínas específicas con funciones concretas y necesarias para el normal funcionamiento del organismo. Un fragmento de la cadena de ADN que contiene las instrucciones para la producción de una proteína concreta es lo que se conoce como gen. Se estima que hay unos 30.000 genes distribuidos en los 46 cromosomas, y cada uno de ellos tiene una localización precisa un cromosoma determinado. Dado que tenemos dos juegos de cromosomas, tenemos dos copias de cada gen, y pasamos a nuestros hijos una de ellas.
La E VHL es una enfermedad de causa genética debida a mutaciones en el gen VHL ubicado en el cromosoma 3 p26-p25 que se puede transmitir de padres a hijos.
Las personas con E VHL tienen una alteración o mutación en la secuencia del gen VHL lo que da lugar a que no se produzca la proteína correspondiente
¿Cómo se hereda o se transmite la VHL?
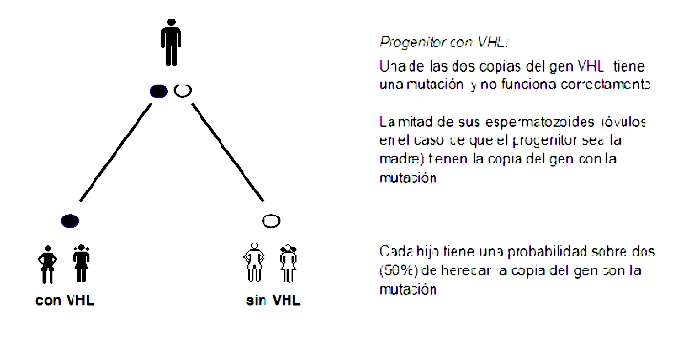
La VHL se hereda siguiendo un patrón dominante. La persona que tiene una mutación en una de las dos copias del gen VHL desarrollará la enfermedad, y cada uno de sus hijos tiene una probabilidad sobre dos (50%) de recibir la copia con la mutación (y la misma probabilidad de recibir la copia sana). Esta probabilidad es la misma en cada gestación y en cada hijo, independientemente del resultado en otro hijo anterior.
En la mayoría de las veces (80%) la VHL se hereda de un progenitor. En el resto de los casos (20%) se trata de una mutación que ocurre por primera vez en el espermatozoide o en el óvulo que ha dado lugar a esa persona como una mutación de “de novo”.
¿Qué riesgo tienen los portadores de desarrollar el síndrome?
La mutación VHL tiene una penetrancia prácticamente completa y los síntomas se desarrollan antes de los 65 años. No se ha observado fenómeno de anticipación, ni tampoco existe una correlación fenotipo-genotipo absoluta. Existe una gran variabilidad en la expresión clínica de la enfermedad.
¿Cómo se diagnostica VHL?
La VHL se puede diagnosticar utilizando los criterios clínicos que requiere la realización de pruebas de bioquímica y de imagen, o bien mediante las pruebas genéticas moleculares del gen VHL.
¿En qué consisten y cuál es la fiabilidad de las pruebas genéticas?
La prueba genética de la VHL consiste en un análisis para detectar mutaciones en el gen MEN1 en el ADN extraído de una muestra de sangre. La probabilidad de encontrar una mutación en una persona con las manifestaciones clínicas típicas de VHL es muy alta y el 72% de las mutaciones se detectan por secuenciación y el 28 % se detectan mediante métodos moleculares que detectan deleciones.
Enfrentarse a la realización de estas pruebas puede tener un impacto emocional y psíquico considerable, por lo que se recomienda que la persona que lo solicite esté convenientemente informada y preparada para ello.
¿Qué debo hacer una vez que conozca el resultado de las pruebas genéticas?
Si se identifica una mutación en el gen VHL en una persona con E VHL, se puede ofrecer la prueba a los familiares cercanos para comprobar si tienen o no la misma mutación en el gen. Aquellos que la tengan desarrollarán la enfermedad y deberán ser atendidos por el especialista correspondiente.
La identificación de los individuos portadores de la mutación en el gen VHL y la vigilancia de las personas que tienen síndrome de VHL incluye el seguimiento mediante pruebas bioquímicas y de imagen evaluación por el oftalmologo y el otorrino.
Por otra parte los individuos no portadores no tendrán que realizar ningún seguimiento particular aunque seguirán presentando un riesgo de cáncer similar al del resto de la población general.
Aquellos familiares que no tengan la mutación pueden tener la seguridad de que no desarrollarán la enfermedad ni la transmitirán a sus hijos.
¿Dónde puedo conseguir más información?
Si necesita más información o desea alguna aclaración adicional, no dude en preguntar a su Médico de Familia, Oncologo o solicitar una nueva visita en la Unidad de Consejo gentico en Cáncer Hereditario.






