La neoplàsia endocrina múltiple tipus 1 (MEN-1), és una síndrome que inclou una combinació de més de 20 tumors endocrins i no endocrins. Els tumors endocrins es fan evidents per la sobreproducció d'hormones pel tumor o pel creixement del tumor en si i inclouen els tumors de paratiroides (en el 90% de les persones entre 20-25 anys d'edat), tumors pituïtaris, tumors endocrins del tracte gastro-enter-pancreàtics (gastrinoma; insulinoma; glucagonoma, VIPoma), tumors carcinoides i tumors adrenocorticales. Els tumors no endocrins associats poden incloure els angiofibromas facials, els colagenomas, els lipomes, els meningiomas, els ependimonas, i els leiomiomas.
Quina és la causa de la MEN-1?
Tota la informació genètica està continguda en el nucli de cadascuna de les nostres cèl·lules, en unes estructures allargades que són els cromosomes. Tenim 46 cromosomes, que realment són dos jocs de 23 cromosomes heretats de cadascun dels nostres pares. Al seu torn, quan tenim fills, els passem a ells, en cadascun dels espermatozoides o òvuls que produïm.
Cada cromosoma és com una madeixa de llana en la qual el fil de la madeixa és la cadena d'ADN que conté la informació genètica. Aquesta informació, emmagatzemada en el codi genètic, consisteix en instruccions per a la producció de proteïnes específiques amb funcions concretes i necessàries per al normal funcionament de l'organisme. Un fragment de la cadena d'ADN que conté les instruccions per a la producció d'una proteïna concreta és el que es coneix com a gen. S'estima que hi ha uns 30.000 gens distribuïts en els 46 cromosomes, i cadascun d'ells té una localització precisa un cromosoma determinat. Atés que tenim dos jocs de cromosomes, tenim dues còpies de cada gen, i passem als nostres fills una d'elles.
La MEN-1 és una malaltia de causa genètica deguda a una alteració o mutació en la seqüència del gen MEN1 situat en el cromosoma 11 el que dóna lloc al fet que no es produïsca la proteïna corresponent.
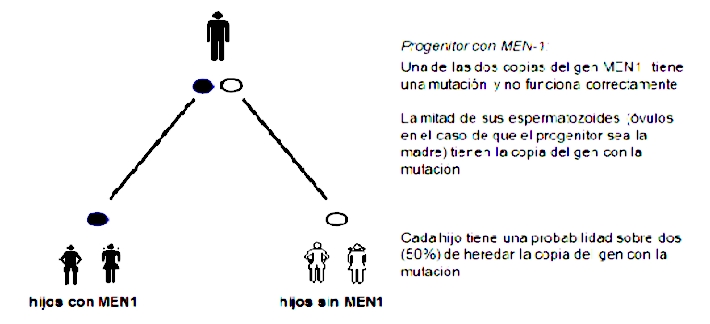
Com s'hereta o es transmet la MEN-1?
La MEN-1 s'hereta seguint un patró dominant. La persona que té una mutació en una de les dues còpies del gen MEN1 desenvoluparà la malaltia, i cadascun dels seus fills té una probabilitat sobre dues (50%) de rebre la còpia amb la mutació (i la mateixa probabilitat de rebre la còpia sana). Aquesta probabilitat és la mateixa en cada gestació i en cada fill, independentment del resultat en un altre fill anterior.
En la majoria de les vegades (90%) la MEN-1 s'hereta d'un progenitor. En la resta dels casos (10%) es tracta d'una mutació que ocorre per primera vegada en l'espermatozoide o en l'òvul i es coneix com una mutació de “de novo”.
Quin risc tenen els portadors de desenvolupar la síndrome?
El portador d'una mutació en el gen MEN1 suposa un risc de desenvolupar en el 100% hiperparatiroidismo primari als 50 anys, sent la primera manifestació en el 90% dels portadors. El risc de desenvolupar tumors de la hipòfisi anterior se situa entre el 10 i 60%, tumors ben diferenciats endocrins entre el 2 i 40%, tumors carcinoides el 10% i tumors *adenocorticales entre el 20 i 40%.
Com es diagnostica la MEN-1?
La MEN-1 es pot diagnosticar utilitzant els criteris clínics de diagnòstic que requereix la realització de proves de bioquímica i d'imatge, o bé mitjançant les proves genètiques moleculars del gen MEN1.
En què consisteixen i quina és la fiabilitat de les proves genètiques?
La prova genètica de la MEN-1 consisteix en una anàlisi per a detectar mutacions en el gen MEN1 en l'ADN extret d'una mostra de sang. La probabilitat de trobar una mutació en una persona amb les manifestacions clíniques típiques de MEN-1 és molt alta i es detecta mutacions en al voltant de 80-90% de familiars afectes de la síndrome de MEN1 i en al voltant del 65% dels individus afectes sense antecedents familiars. En la resta dels casos, no trobar una mutació pot deure's a diverses causes (limitacions tècniques de la pròpia anàlisi, la possible participació d'un altre gen, conegut o no, etc.), i NO descarta forçosament el diagnòstic de MEN1.
Enfrontar-se a la realització d'aquestes proves pot tindre un impacte emocional i psíquic considerable, per la qual cosa es recomana que la persona que el sol·licite estiga convenientment informada i preparada per a això.
Què he de fer una vegada que conega el resultat de les proves genètiques?
Si s'identifica una mutació en el gen MEN1 en una persona afecta, es pot oferir la prova als familiars propers per a comprovar si tenen o no la mateixa mutació en el gen. Aquells que la tinguen desenvoluparan la malaltia i hauran de ser atesos per l'especialista corresponent.
La identificació dels individus portadors de la mutació en el gen MEN1 i la vigilància de les persones que tenen síndrome de MEN1 inclou el seguiment mitjançant proves bioquímiques (concentracions sèriques de calci, de gastrina, de polipèptid pancreàtic i de prolactina) i proves d'imatge abdominal o del cap.
D'altra banda els individus no portadors no desenvoluparan la malaltia, ni la transmetran als seus fills i no hauran de realitzar cap seguiment particular. Aquestes persones presenten un risc de càncer similar a la població general i han de realitzar les mesures de detecció precoç per a la prevenció del càncer aplicables per a la població general en el nostre medio.en ocasions pot resultar difícil d'interpretar l'alteració detectada (no totes les mutacions són iguals) perquè no es coneix en el moment actual si està associada a un augment de risc de desenvolupar MEN1.
On puc aconseguir més informació?
Si necessita més informació o desitja algun aclariment addicional, no dubte a preguntar al seu Metge de Família, Oncòlogo o sol·licitar una nova visita en la Unitat de Consell genètic en Càncer Hereditari.






